

Rac1 promotes kidney collecting duct integrity by limiting actomyosin activity
- PMID: 34647970
- PMCID: PMC8563289
- DOI: 10.1083/jcb.202103080
Rac1 promotes kidney collecting duct integrity by limiting actomyosin activity
Abstract
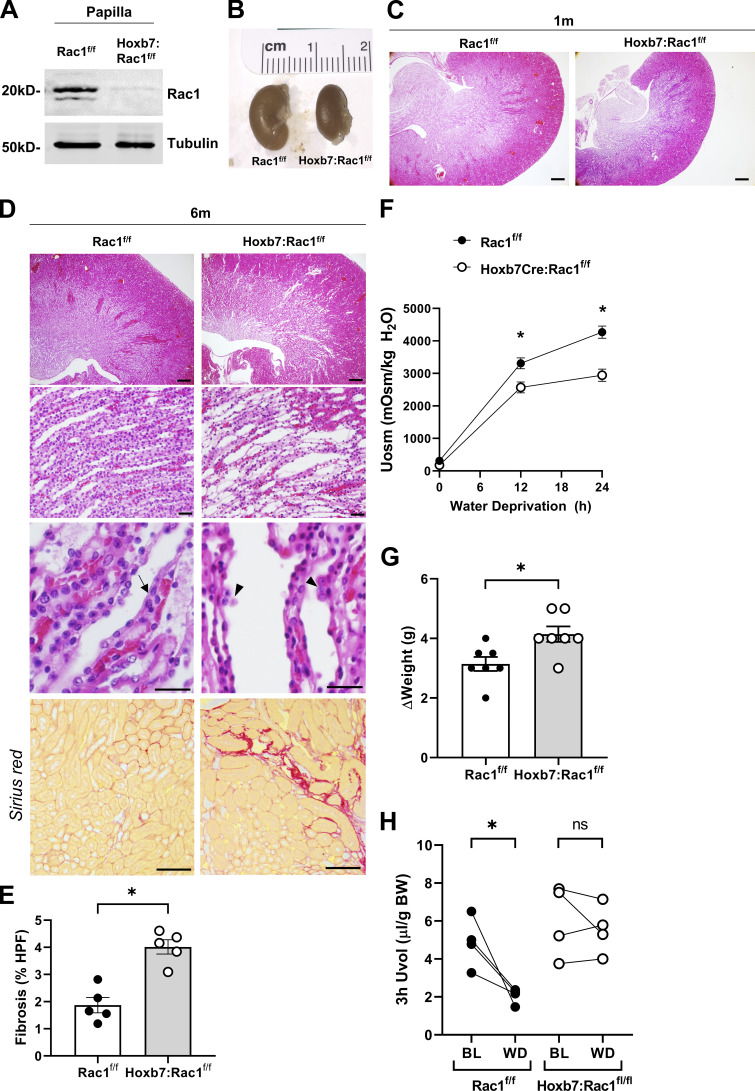
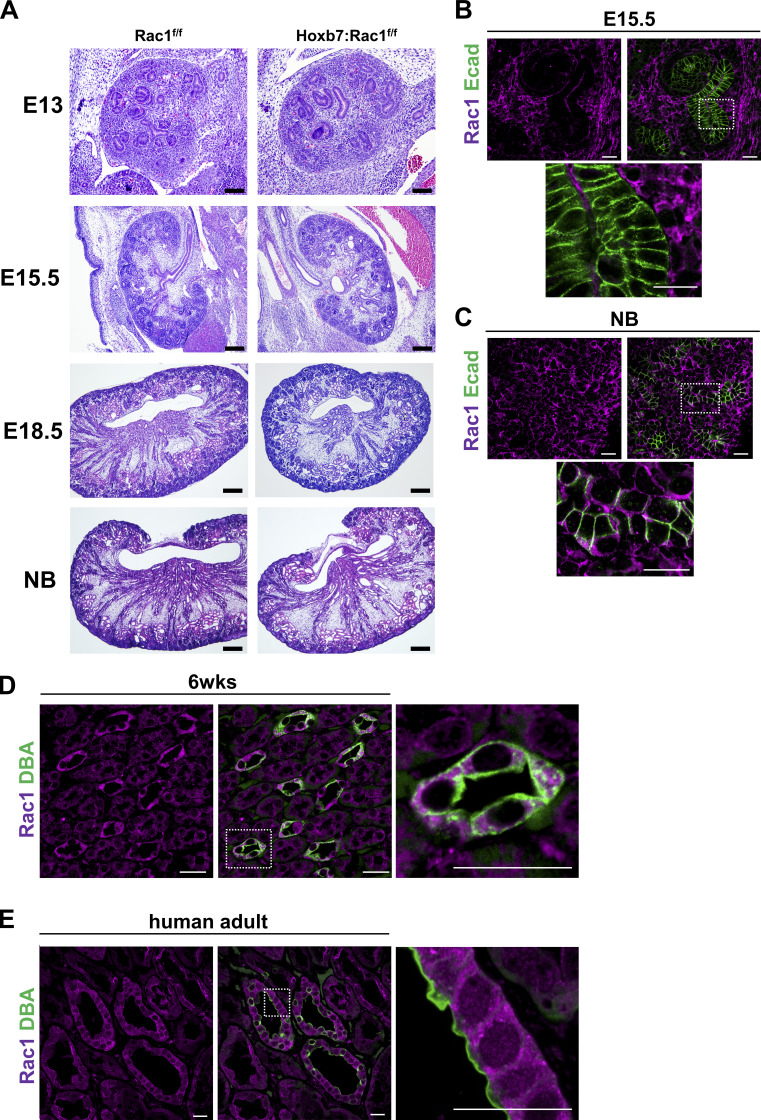
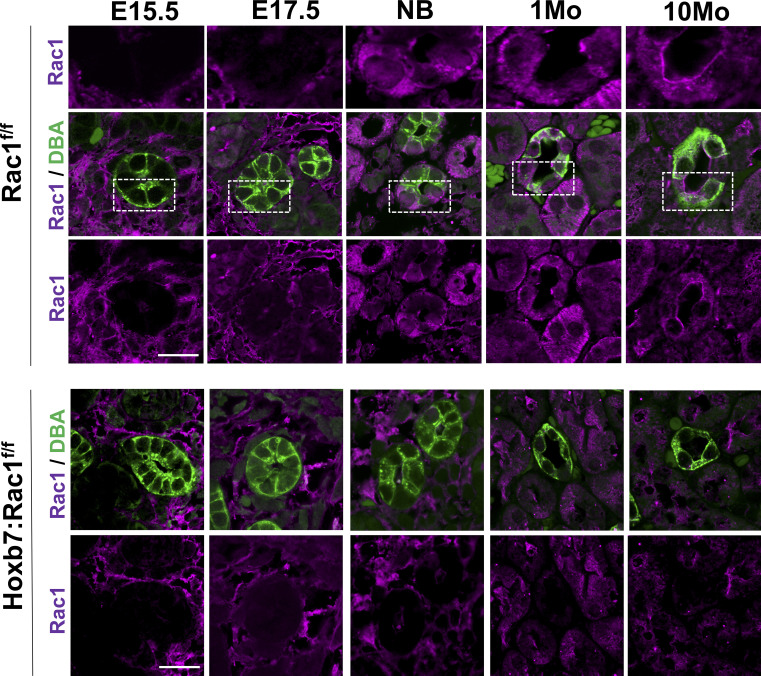
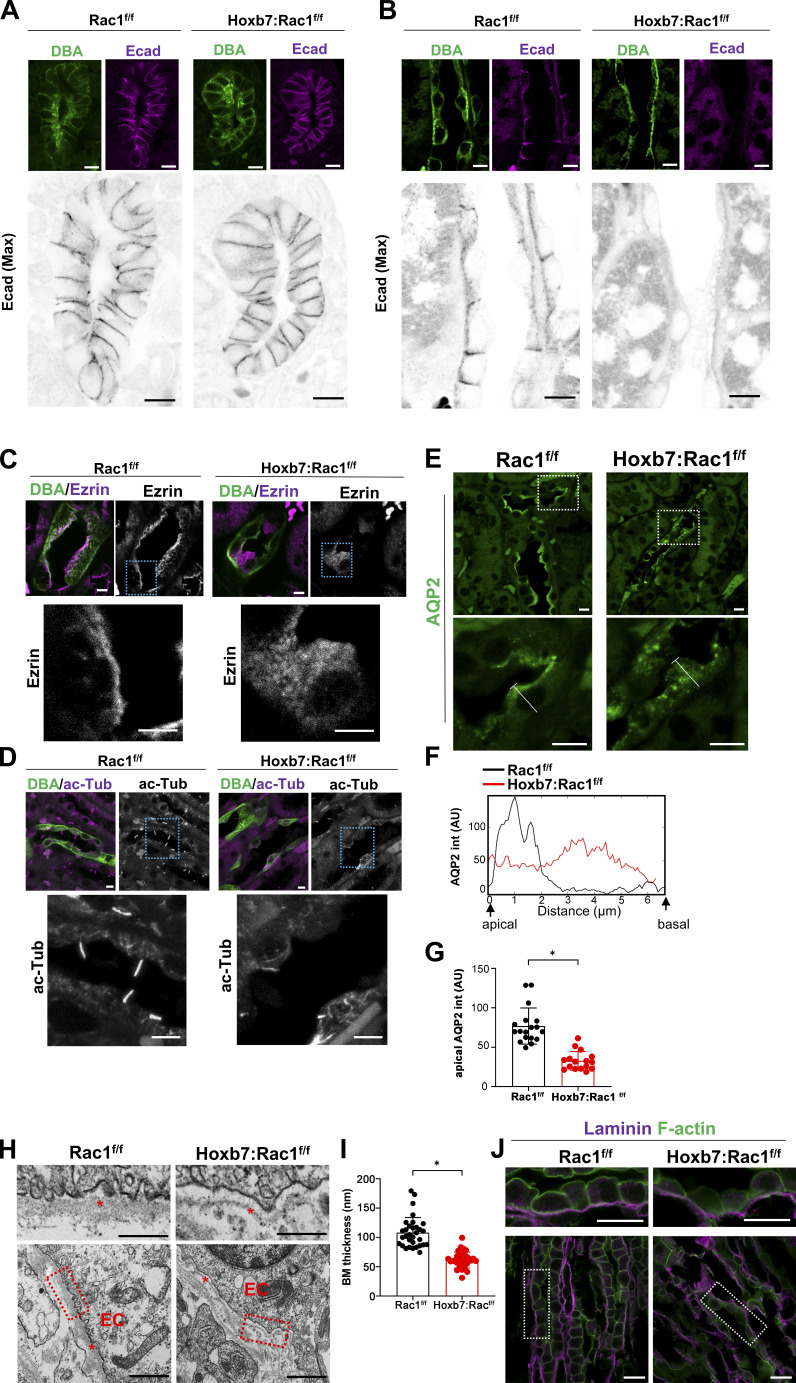
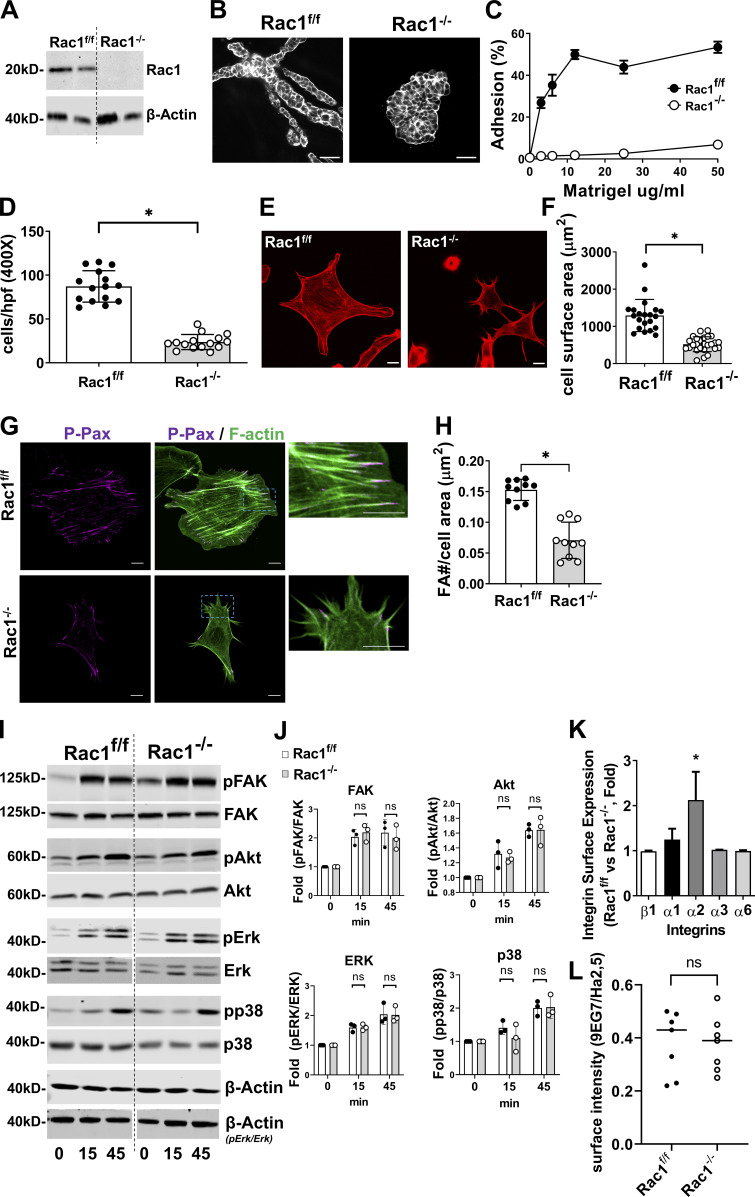
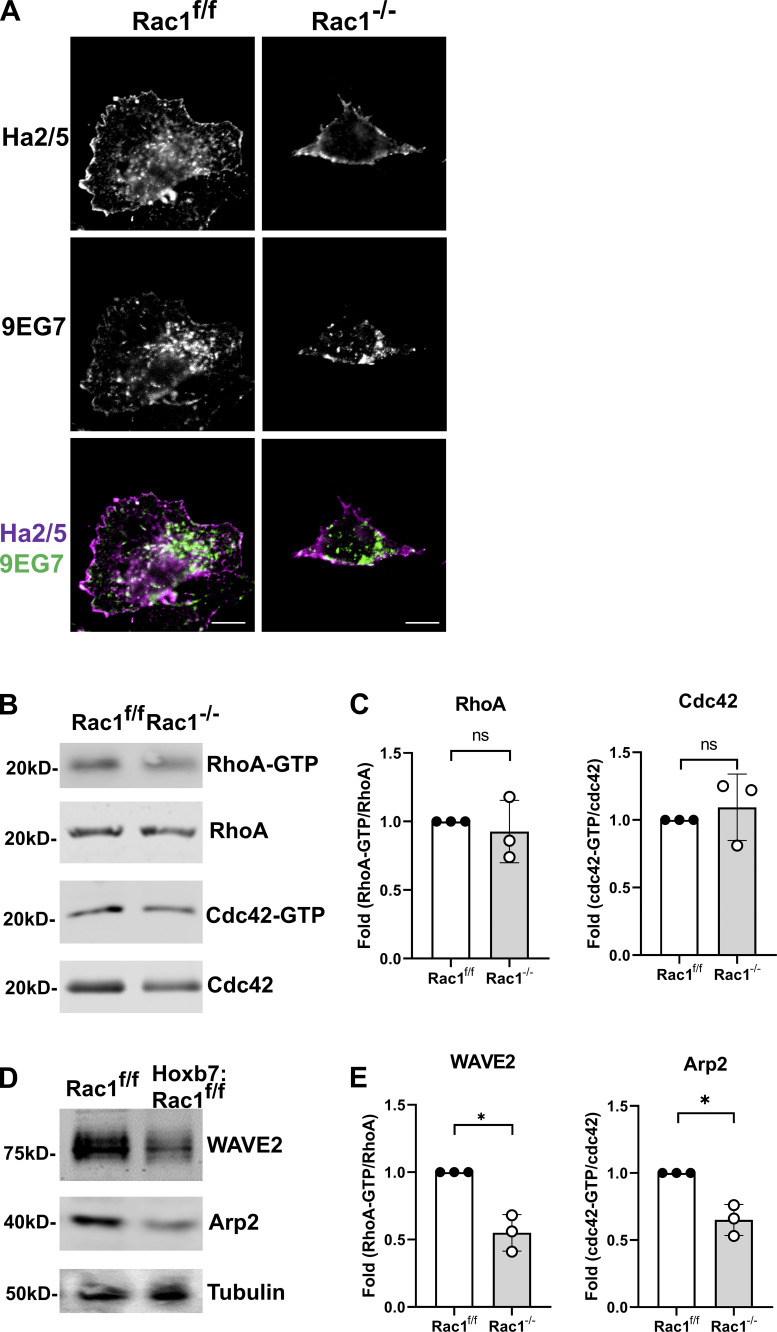
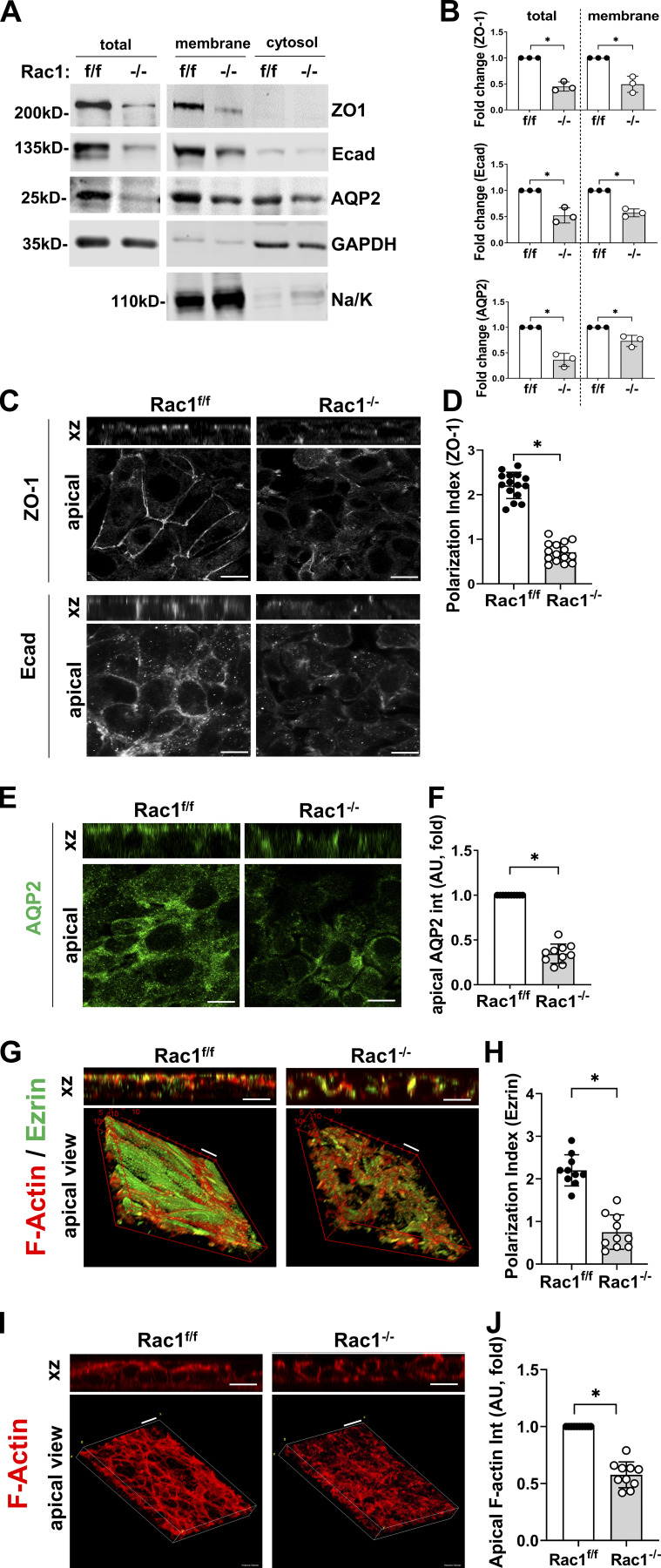
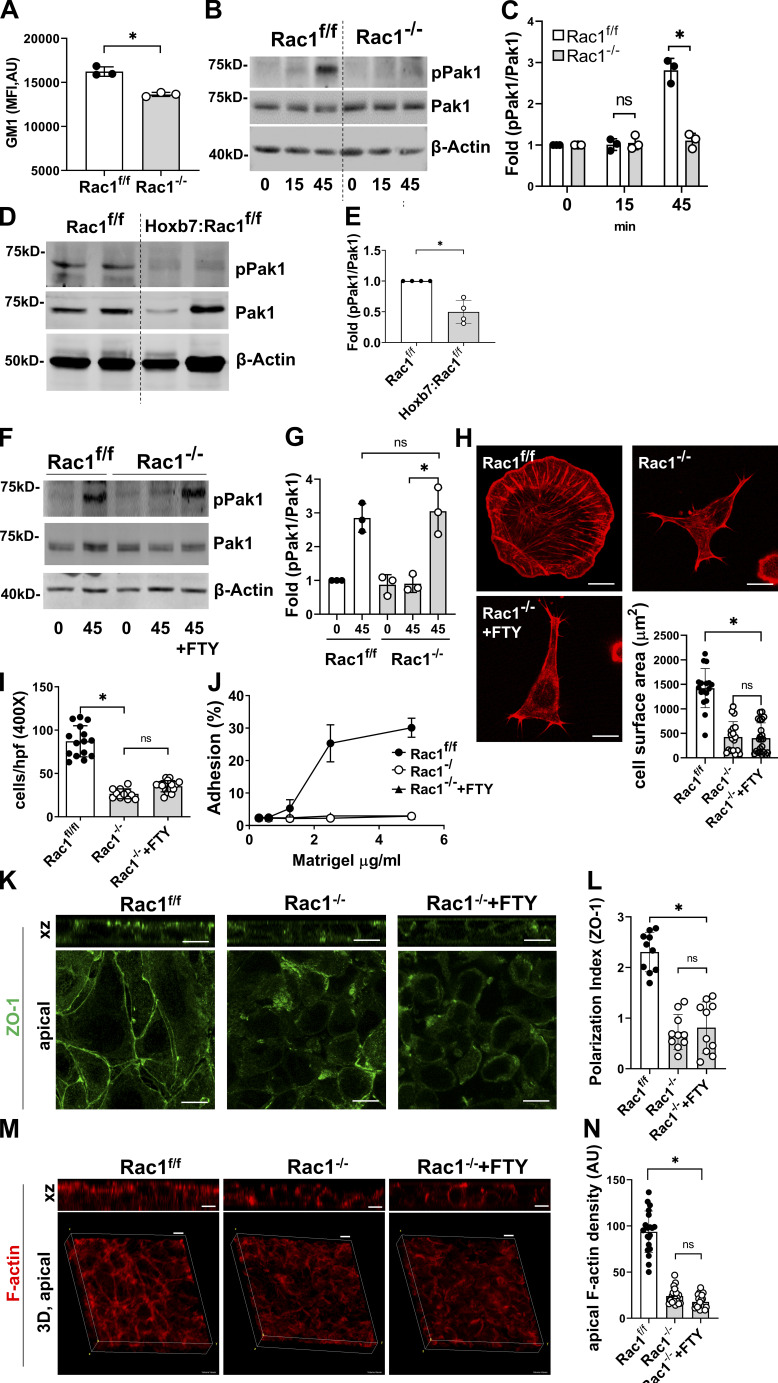
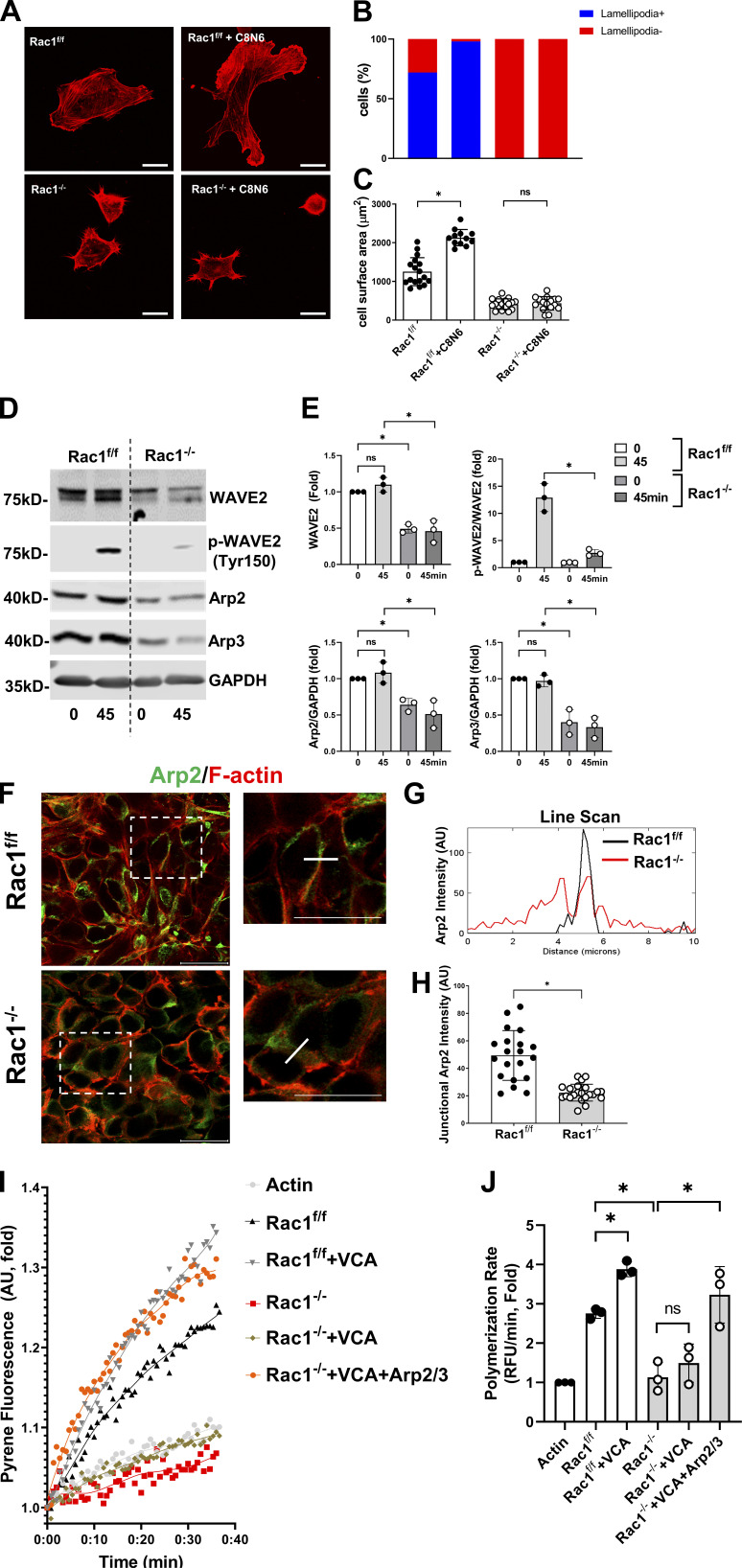
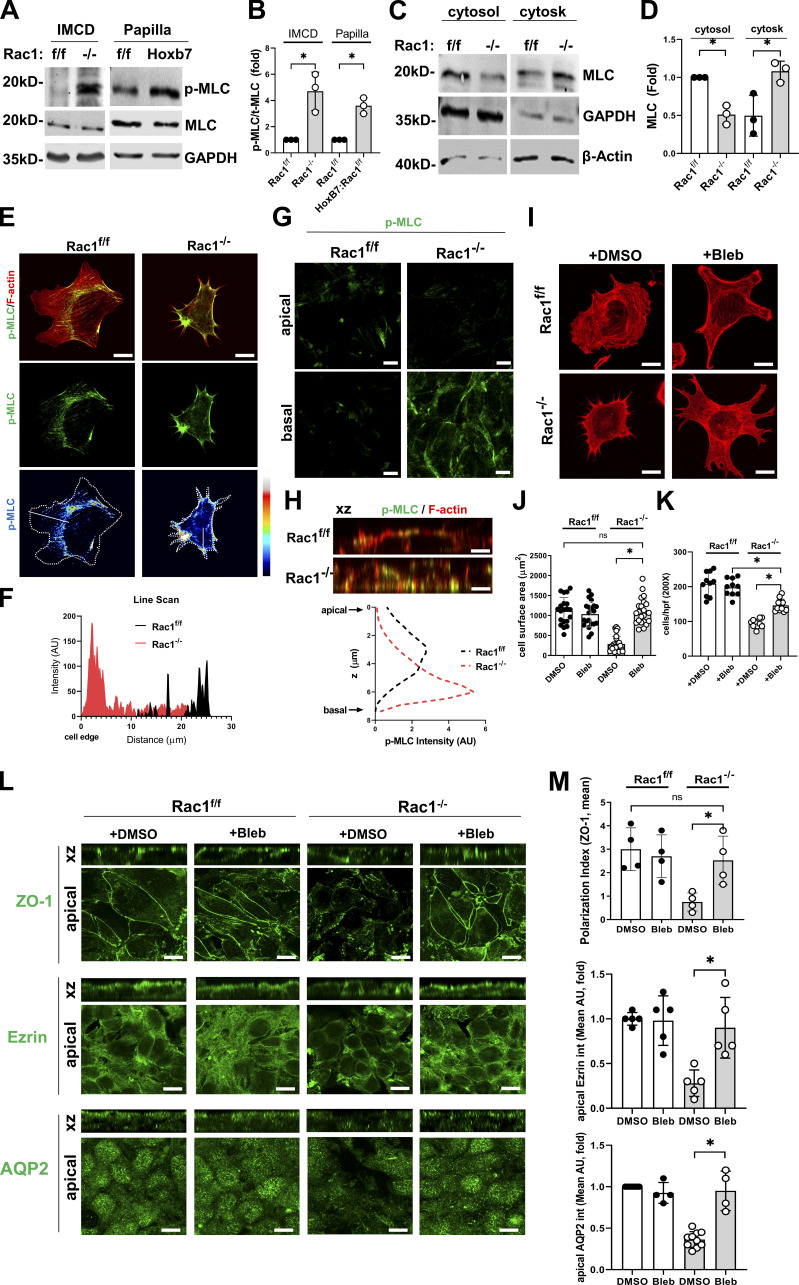
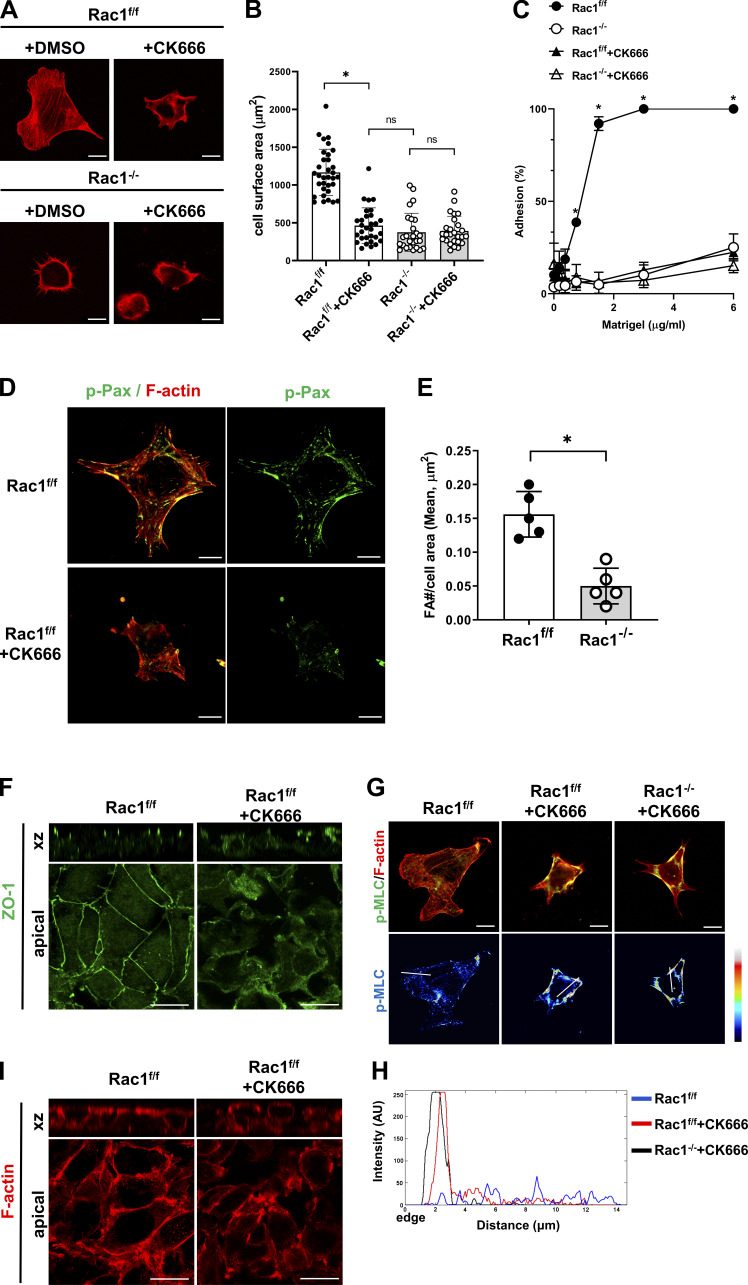
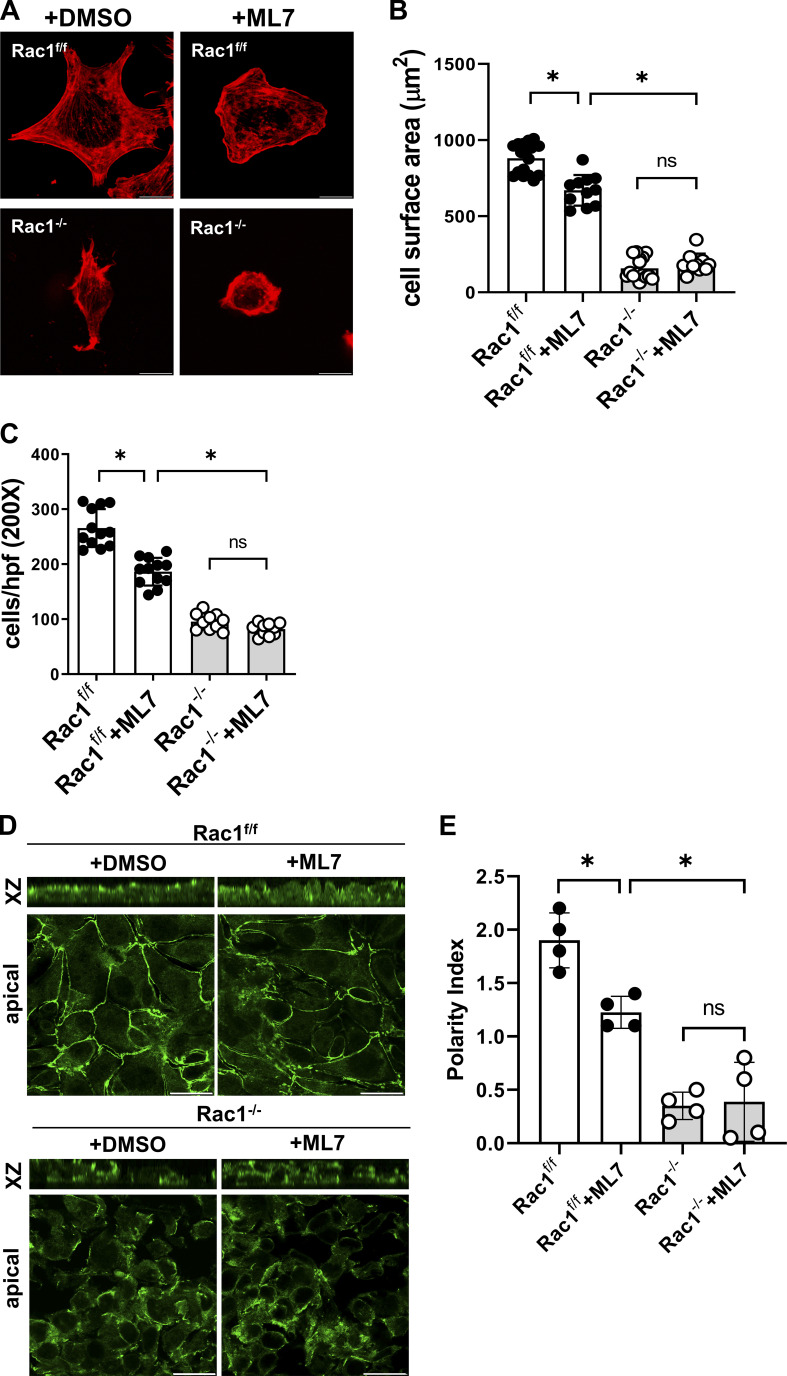
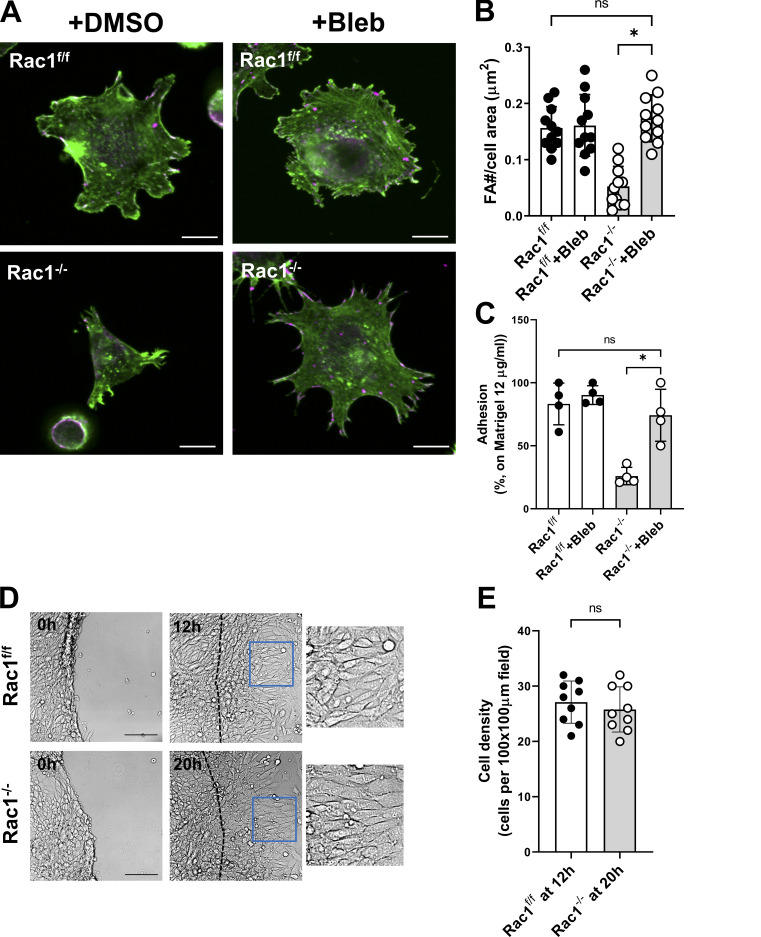
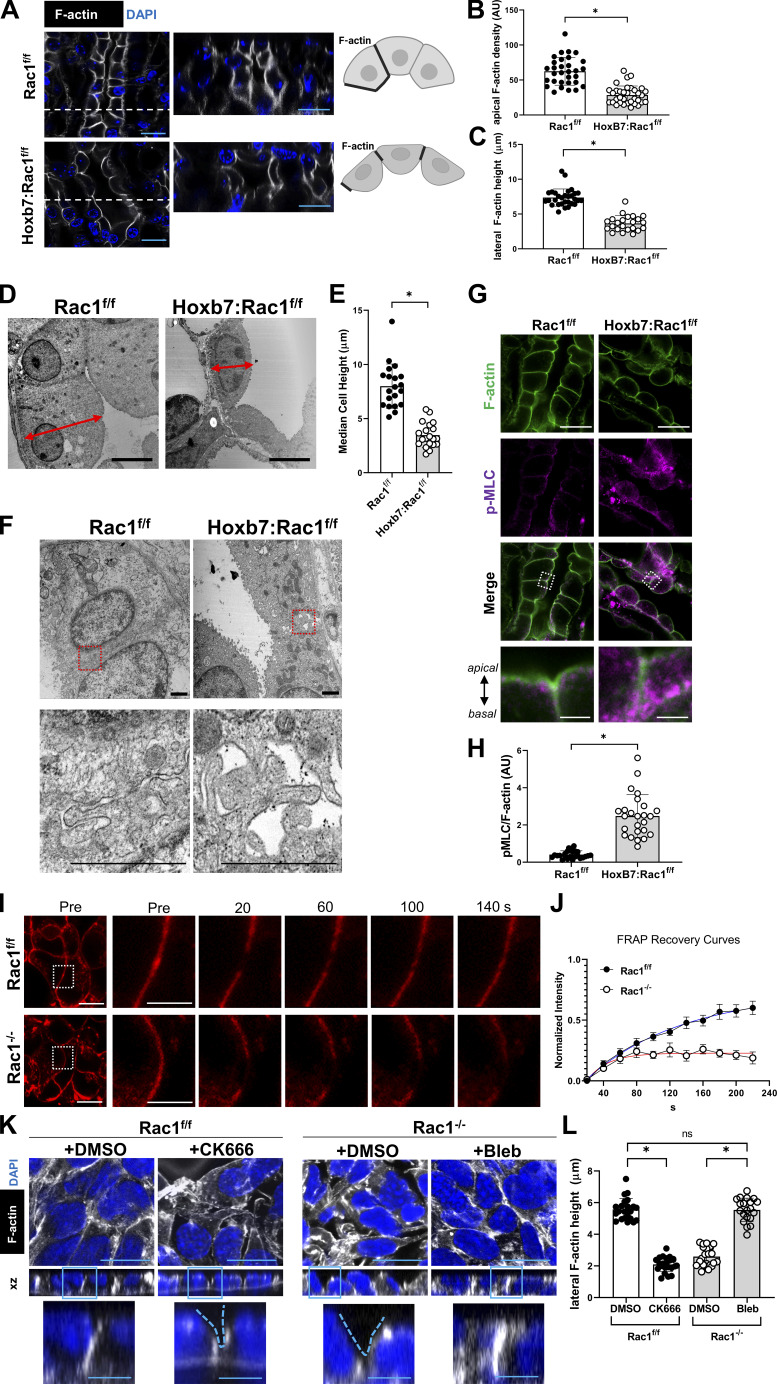
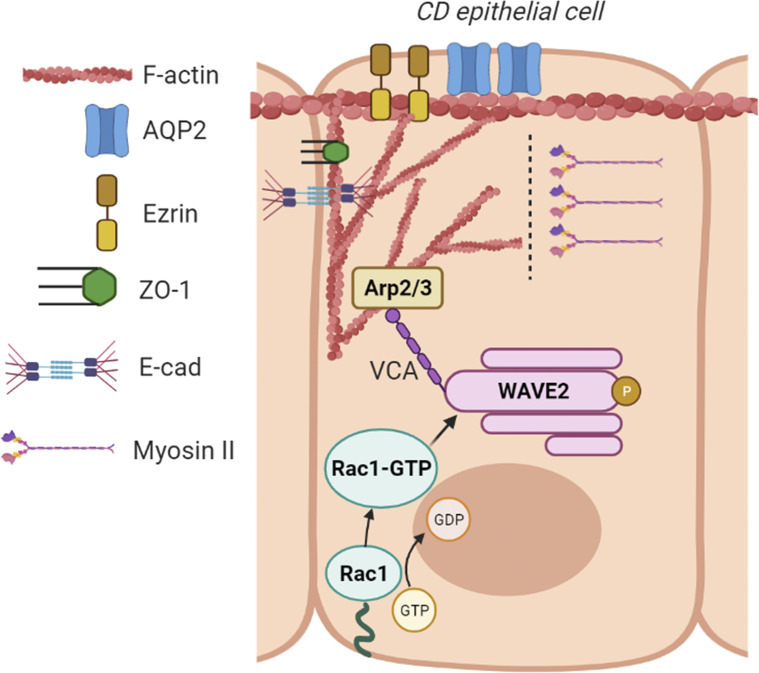
A polarized collecting duct (CD), formed from the branching ureteric bud (UB), is a prerequisite for an intact kidney. The small Rho GTPase Rac1 is critical for actin cytoskeletal regulation. We investigated the role of Rac1 in the kidney collecting system by selectively deleting it in mice at the initiation of UB development. The mice exhibited only a mild developmental phenotype; however, with aging, the CD developed a disruption of epithelial integrity and function. Despite intact integrin signaling, Rac1-null CD cells had profound adhesion and polarity abnormalities that were independent of the major downstream Rac1 effector, Pak1. These cells did however have a defect in the WAVE2-Arp2/3 actin nucleation and polymerization apparatus, resulting in actomyosin hyperactivity. The epithelial defects were reversible with direct myosin II inhibition. Furthermore, Rac1 controlled lateral membrane height and overall epithelial morphology by maintaining lateral F-actin and restricting actomyosin. Thus, Rac1 promotes CD epithelial integrity and morphology by restricting actomyosin via Arp2/3-dependent cytoskeletal branching.
© 2021 Bock et al.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- I01 BX000320/BX/BLRD VA/United States
- R01 DK069921/DK/NIDDK NIH HHS/United States
- T32 DK007569/DK/NIDDK NIH HHS/United States
- R01-DK069921/NH/NIH HHS/United States
- P30 DK058404/DK/NIDDK NIH HHS/United States
- 5P30 CA68485-19/NH/NIH HHS/United States
- S10 OD021630/OD/NIH HHS/United States
- R01 DK108968/DK/NIDDK NIH HHS/United States
- I01 BX002025/BX/BLRD VA/United States
- CA68485/NH/NIH HHS/United States
- DK058404/NH/NIH HHS/United States
- R01 DK119212/DK/NIDDK NIH HHS/United States
- P30 EY008126/EY/NEI NIH HHS/United States
- 1I01BX002196/VA/VA/United States
- P30 CA068485/CA/NCI NIH HHS/United States
- P30 CA68485/NH/NIH HHS/United States
- R01 DK056942/DK/NIDDK NIH HHS/United States
- U24 DK059637/DK/NIDDK NIH HHS/United States
- IK6 BX005240/BX/BLRD VA/United States
- I01 BX002196/BX/BLRD VA/United States
- R01 DK127589/DK/NIDDK NIH HHS/United States
- S10-OD021630/NH/NIH HHS/United States
- P30 DK020593/DK/NIDDK NIH HHS/United States
- I01 BX003425/BX/BLRD VA/United States
- AHA/American Heart Association-American Stroke Association/United States
- P30 DK114809/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
